Research
AI, structural bioinformatics, membrane proteins, protein interactions, and evolution.
Our research investigates how proteins are built, how they interact, how they evolve, and how to model these processes using modern computational methods. We work at the interface of bioinformatics, machine learning, structural biology, and evolutionary analysis, with a strong focus on method development that enables new biological insight.
AI and structural modelling of protein interactions
Proteins rarely act alone. A central goal in the lab is to predict how proteins recognize one another and how larger assemblies are built from pairwise and higher-order interactions. We develop methods that use AlphaFold-based modelling and related machine-learning strategies to predict protein-protein interactions and reconstruct large complexes. Current work extends this toward proteome-scale interaction mapping and structural models of molecular systems in human cells.
Proteoforms and the molecular language of the cell
A gene does not produce a single invariant product. Splicing and post-translational modifications generate many proteoforms, and these different molecular states shape cellular function. In our current work, we combine long-read transcriptomics, proteomics, deep learning, and structural modelling to identify proteoforms, predict their interaction partners, and build atomistic models of the resulting complexes. The long-term goal is a more complete and mechanistic description of the human proteomic landscape.
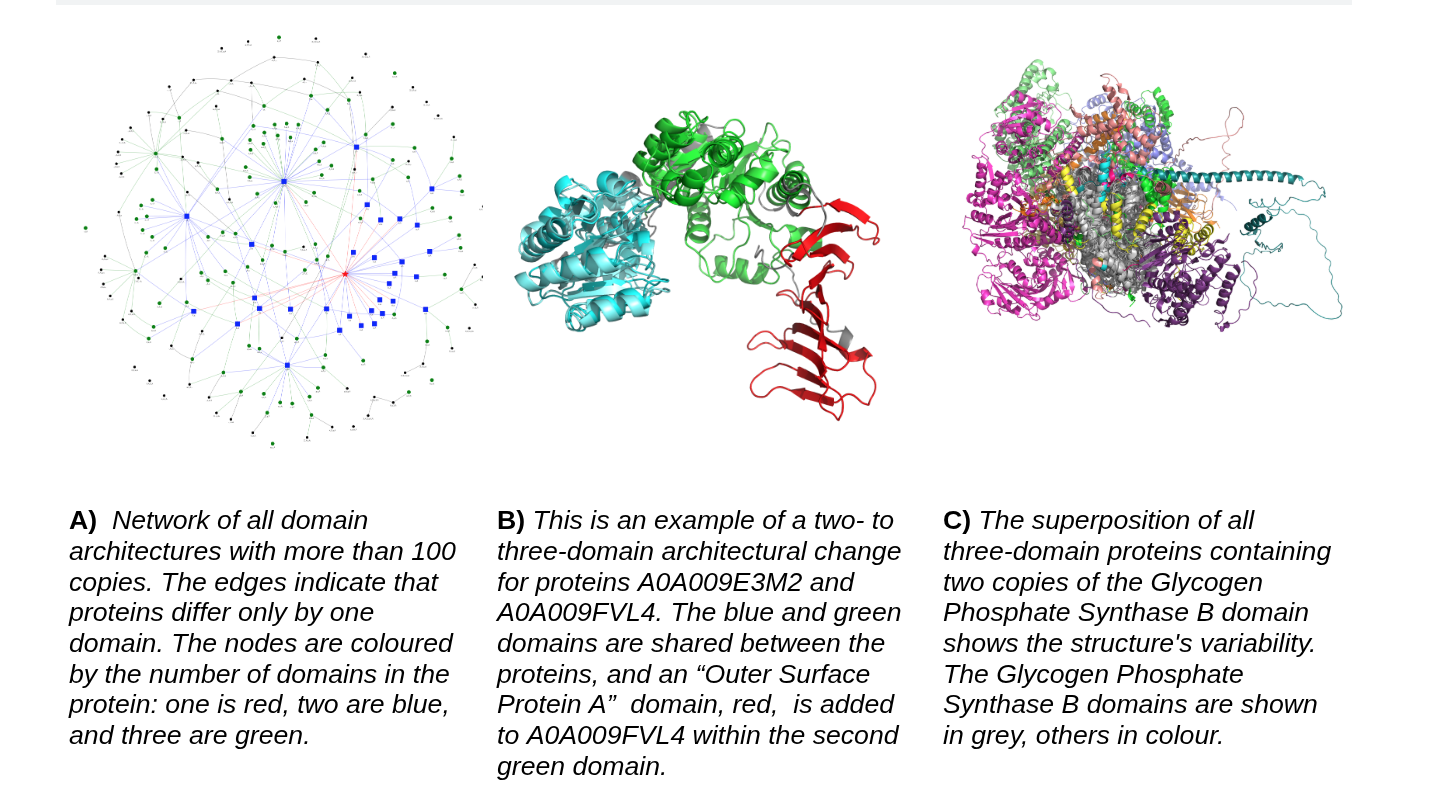
Protein evolution and domain architecture
Proteins evolve under structural and functional constraints. We are interested in how domains change, how new multi-domain architectures arise, and how structural diversification shapes protein families across evolution. With modern resources such as AlphaFoldDB and TED, these questions can now be studied at a scale that was previously impossible.
Membrane proteins and topology
The lab has a long-standing interest in membrane proteins and the sequence-structure principles that govern their insertion, topology, and folding. This includes prediction of transmembrane helices, topology constraints, and the structural organization of membrane proteins. These themes remain important both biologically and methodologically, and they continue to influence how we think about protein architecture and evolution.
Current funded work includes large collaborative efforts on proteoforms and molecular interactions together with projects on protein domain architecture evolution. We are especially interested in problems where new computational ideas unlock previously inaccessible biology, and in building methods that are both accurate and interpretable so computational advances translate into mechanistic understanding.