Sequence alignments
- http://en.wikipedia.org/wiki/Sequence_alignment
- http://en.wikipedia.org/wiki/Needleman-Wunsch_algorithm
- http://en.wikipedia.org/wiki/Substitution_matrix
- http://en.wikipedia.org/wiki/BLOSUM
- http://en.wikipedia.org/wiki/Point_accepted_mutation
Extra information
- http://www.avatar.se/molbioinfo2001/dynprog/dynamic.html
- http://en.wikipedia.org/wiki/Smith%E2%80%93Waterman_algorithm
- Lambert_review_2003
Slides
- Sequence Alignment
 Loading...
Loading...
Videos
- https://youtu.be/nbj9RncHg7o
- https://youtu.be/dnG_1-5XJ50
- https://www.youtube.com/watch?v=PZJiHIwNblU
- https://youtu.be/sSHl_23hToM
- https://www.youtube.com/watch?v=jTfGPzRj0s8
- Discussion session about Sequence alignments
Good lectures by others
- https://www.youtube.com/watch?v=-KQ6VtOTfzw
- https://www.youtube.com/watch?v=vqxc2EfPWdk
- https://www.youtube.com/watch?v=zwA-6_1bLgE
Old Videos
Dynamic programming (from Per Kraulis avatar.se)
Dynamic Programming
The following is an example of global sequence alignment using Needleman/Wunsch techniques. For this example, the two sequences to be globally aligned are
G A A T T C A G T T A (sequence #1)
G G A T C G A (sequence #2)
So M = 11 and N = 7 (the length of sequence #1 and sequence #2, respectively)
A simple scoring scheme is assumed where
- Si,j = 1 if the residue at position i of sequence #1 is the same as the residue at position j of sequence #2 (match score); otherwise
- Si,j = 0 (mismatch score)
- w = 0 (gap penalty)
Three steps in dynamic programming
- Initialization
- Matrix fill (scoring)
- Traceback (alignment)
Since this example assumes there is no gap opening or gap extension penalty, the first row and first column of the matrix can be initially filled with 0.
For each position, Mi,j is defined to be the maximum score at position i,j; i.e.
Mi,j = MAXIMUM[
Mi-1, j-1 + Si,j (match/mismatch in the diagonal),
Mi,j-1 + w (gap in sequence #1),
Mi-1,j + w (gap in sequence #2)]
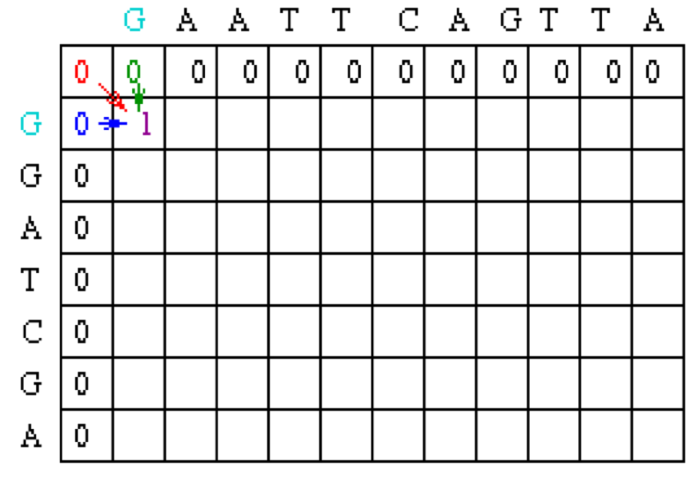
Note that in the example, Mi-1,j-1 will be red, Mi,j-1 will be green and Mi-1,j will be blue.
Using this information, the score at position 1,1 in the matrix can be calculated. Since the first residue in both sequences is a G, S1,1 = 1, and by the assumptions stated at the beginning, w = 0. Thus, M1,1 = MAX[M0,0 + 1, M1, 0 + 0, M0,1 + 0] = MAX [1, 0, 0] = 1.
A value of 1 is then placed in position 1,1 of the scoring matrix.
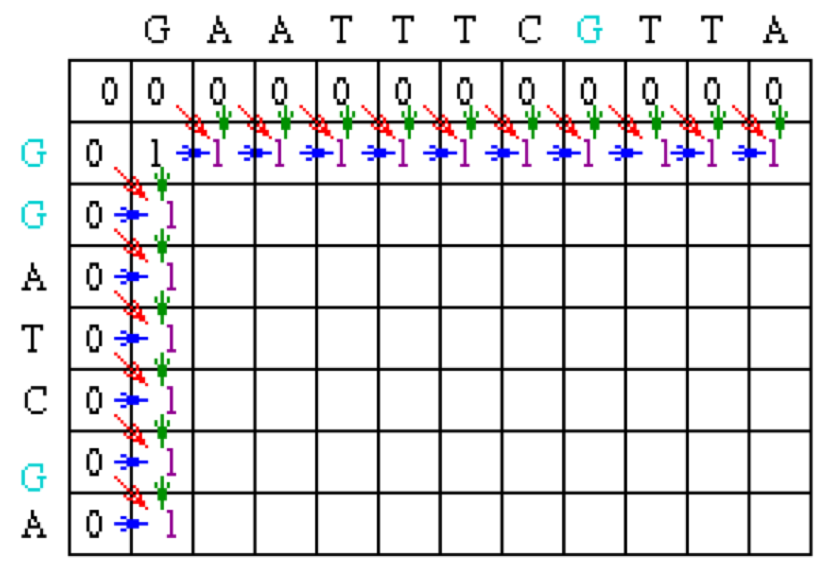
Since the gap penalty (w) is 0, the rest of row 1 and column 1 can be filled in with the value 1. Take the example of row 1. At column 2, the value is the max of 0 (for a mismatch), 0 (for a vertical gap) or 1 (horizontal gap). The rest of row 1 can be filled out similarly until we get to column 8. At this point, there is a G in both sequences (light blue). Thus, the value for the cell at row 1 column 8 is the maximum of 1 (for a match), 0 (for a vertical gap) or 1 (horizontal gap). The value will again be 1. The rest of row 1 and column 1 can be filled with 1 using the above reasoning.
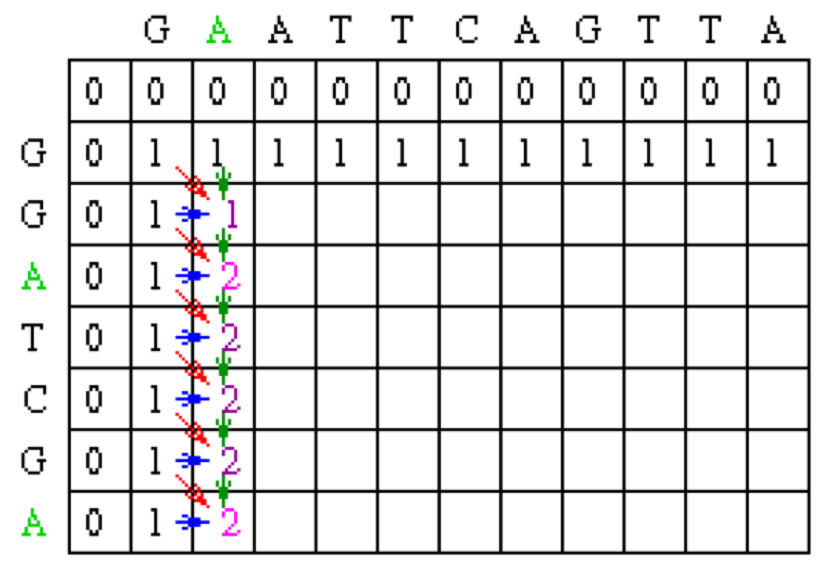
Now let’s look at column 2. The location at row 2 will be assigned the value of the maximum of 1(mismatch), 1(horizontal gap) or 1 (vertical gap). So its value is 1.
At the position column 2 row 3, there is an A in both sequences. Thus, its value will be the maximum of 2(match), 1 (horizontal gap), 1 (vertical gap) so its value is 2.
Moving along to position colum 2 row 4, its value will be the maximum of 1 (mismatch), 1 (horizontal gap), 2 (vertical gap) so its value is 2. Note that for all of the remaining positions except the last one in column 2, the choices for the value will be the exact same as in row 4 since there are no matches. The final row will contain the value 2 since it is the maximum of 2 (match), 1 (horizontal gap) and 2(vertical gap).
Using the same techniques as described for column 2, we can fill in column 3.
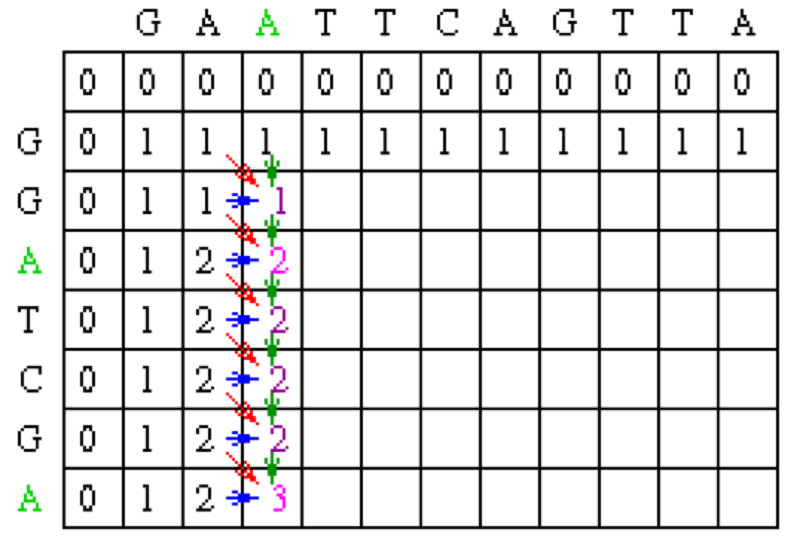
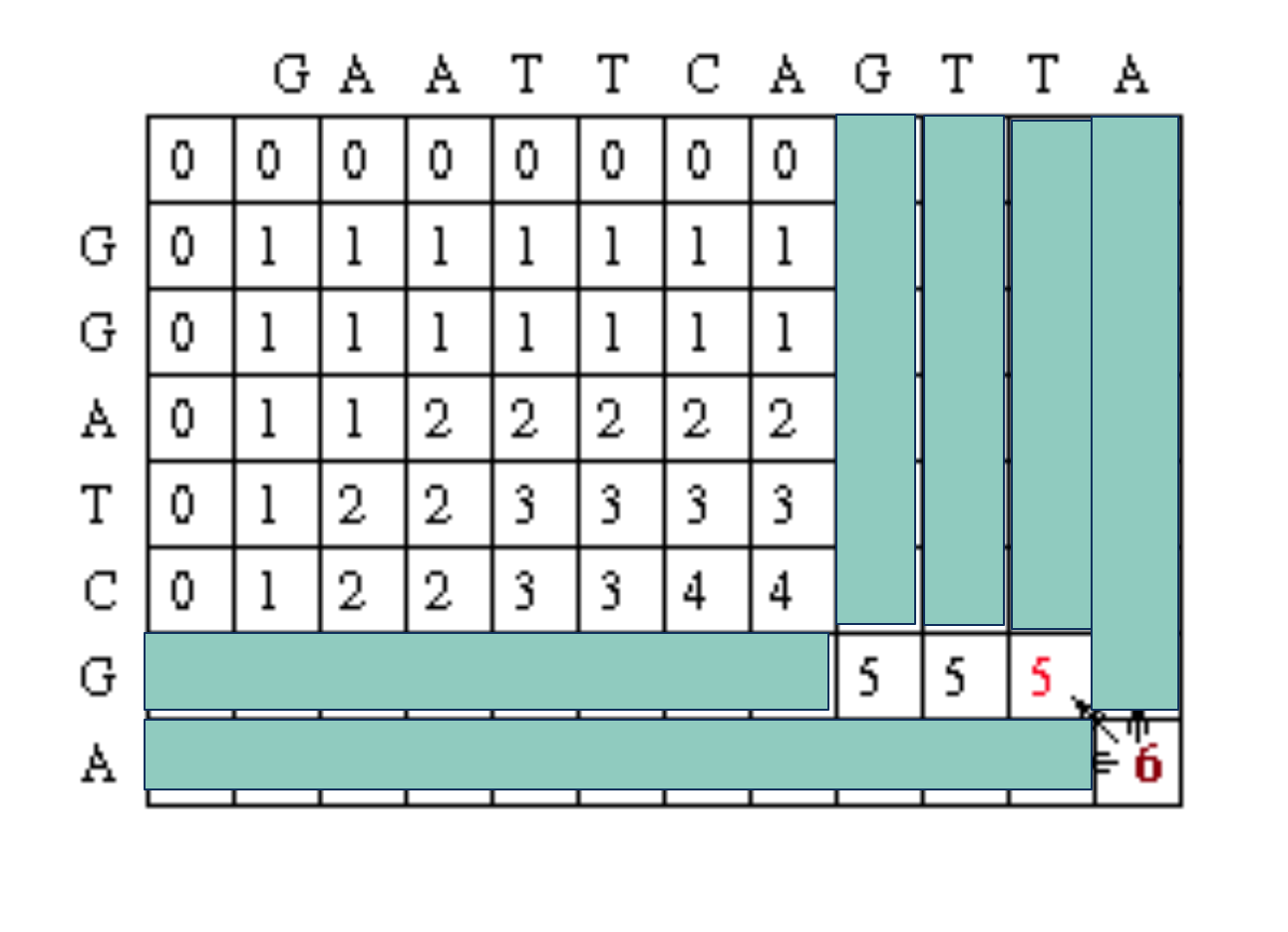
After filling in all of the values the score matrix is as follows:
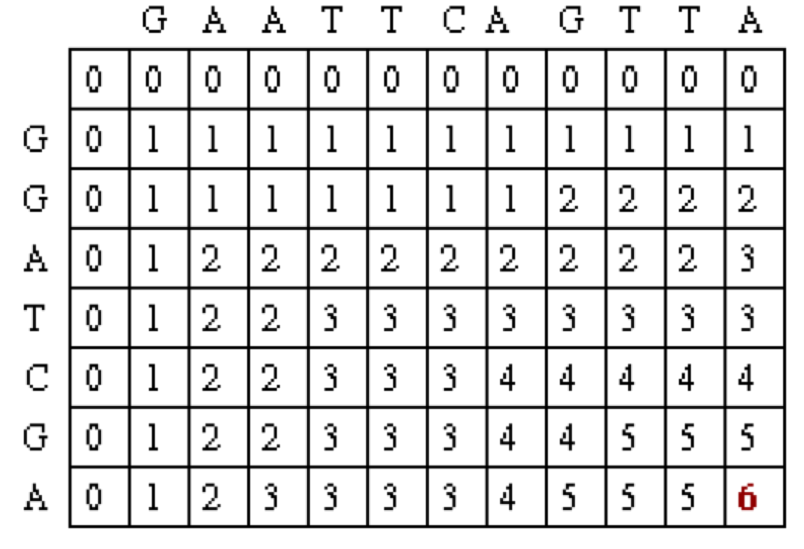
The traceback step begins in the M,J position in the matrix, i.e. the position that leads to the maximal score. In this case, there is a 6 in that location.
Traceback takes the current cell and looks to the neighbor cells that could be direct predacessors. This means it looks to the neighbor to the left (gap in sequence #2), the diagonal neighbor (match/mismatch), and the neighbor above it (gap in sequence #1). The algorithm for traceback chooses as the next cell in the sequence one of the possible predacessors. In this case, the neighbors are marked in red. They are all also equal to 5.
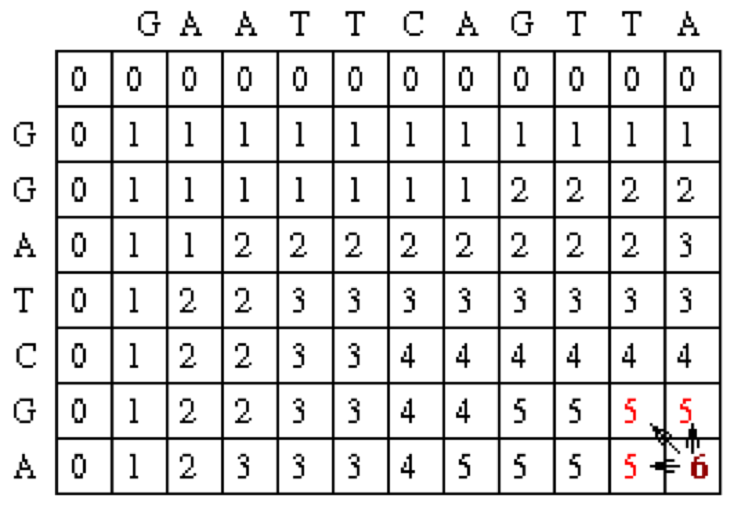
Since the current cell has a value of 6 and the scores are 1 for a match and 0 for anything else, the only possible predecessor is the diagonal match/mismatch neighbor. If more than one possible predecessor exists, any can be chosen. This gives us a current alignment of
(Seq #1) A
|
(Seq #2) A
So now we look at the current cell and determine which cell is its direct predecessor. In this case, it is the cell with the red 5.
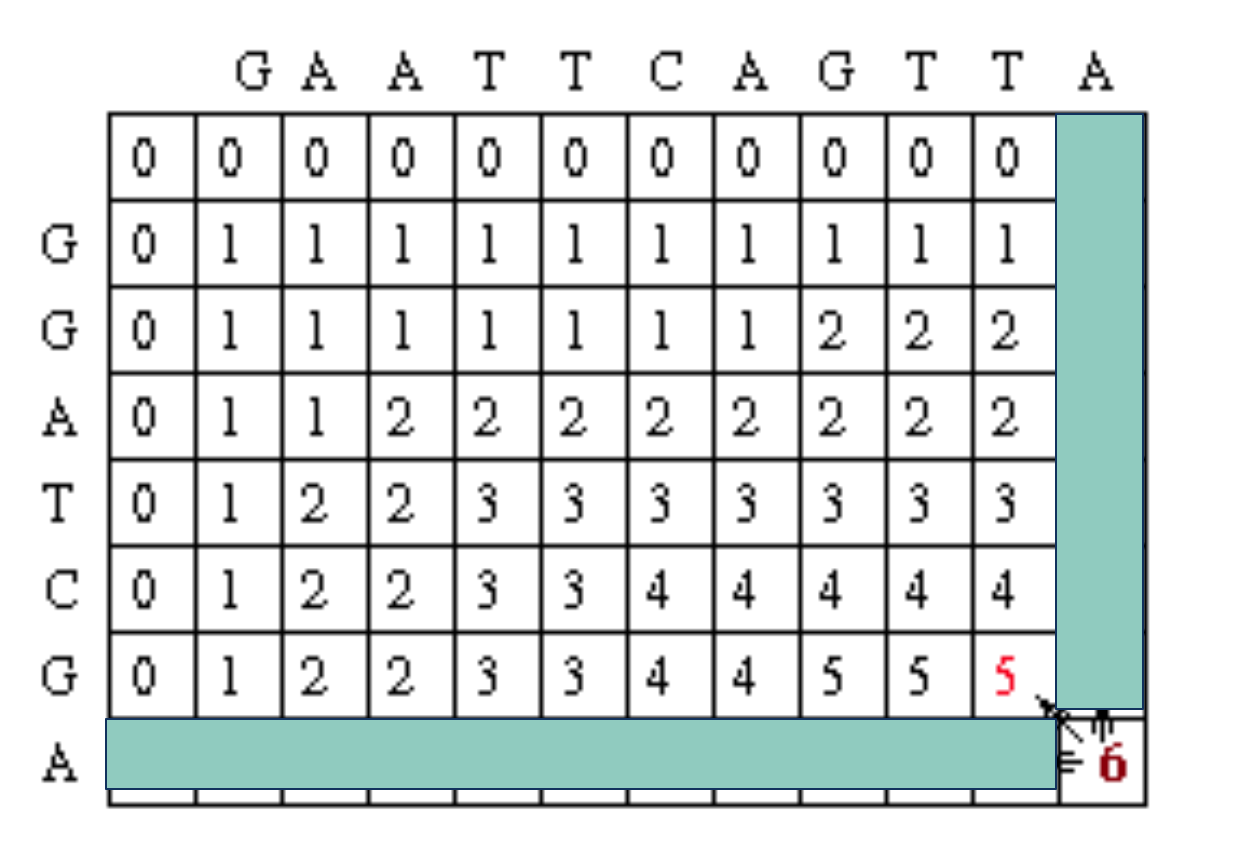
The alignment as described in the above step adds a gap to sequence #2, so the current alignment is
(Seq #1) T A
|
(Seq #2) _ A
Once again, the direct predecessor produces a gap in sequence #2.
(Seq #1) T T A
|
_ _ A
Continuing on with the traceback step, we eventually get to a position in column 0 row 0 which tells us that traceback is completed. One possible maximum alignment is :
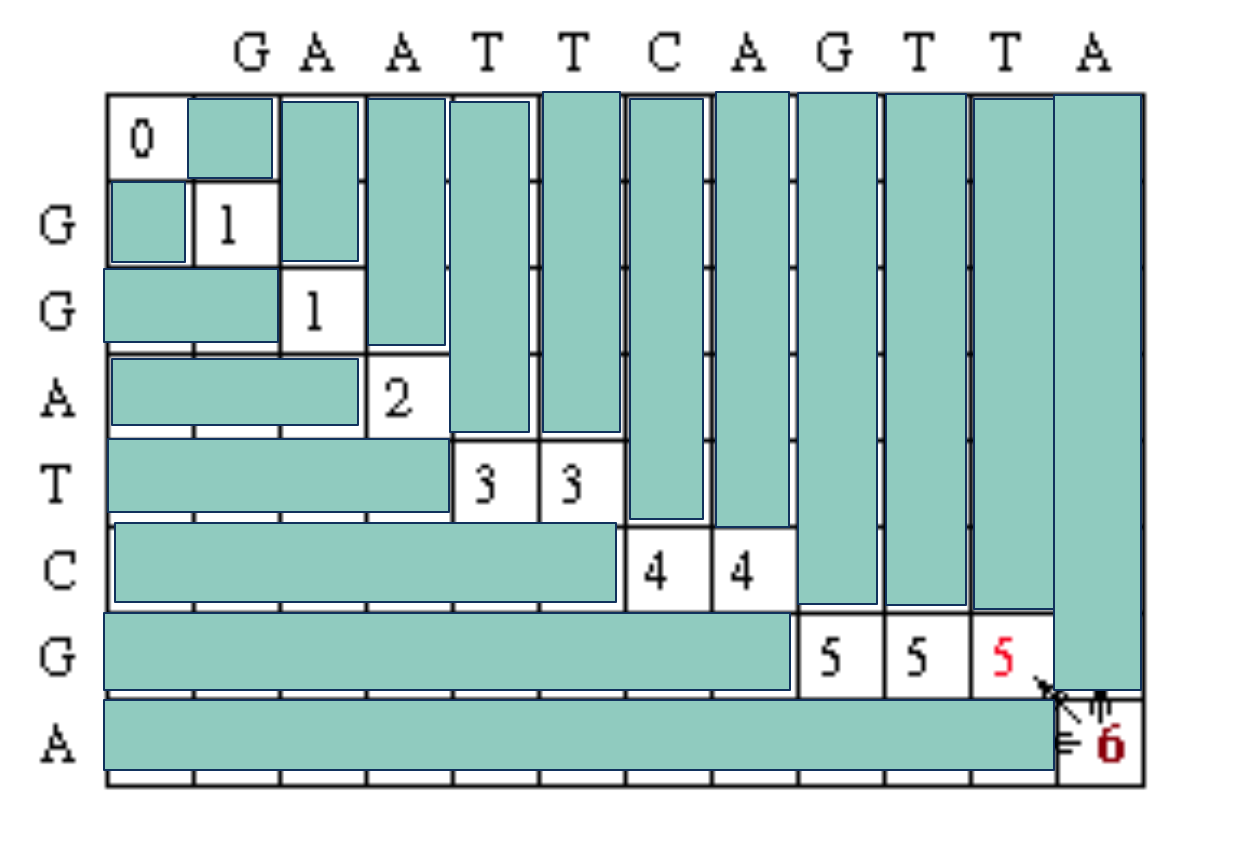
G A A T T C A G T T A
| | | | | |
G G A _ T C _ G _ _ A
An alternate solution is:
G _ A A T T C A G T T A
| | | | | |
G G _ A _ T C _ G _ _ A
There are more alternative solutions each resulting in a maximal global alignment score of 6. Since this is an exponential problem, most dynamic programming algorithms will only print out a single solution.
Now you are ready to explore an example using an advanced scoring scheme.
1. If PAM is not a scoring matrix, what is the outcome? Is it used to show whether two sequences come from distant or closer relatives? Then when wanting to compare two sequences of two different organisms, do we have to know in advance how related they might be, so we chose the right PAM? On the same aspect, how do we know which BLOSUM to use, and what is exactly the outcome, if not a score?
2. Concerning global and local alignment, does local still requires a full comparison of two sequences, but it just returns the most conserved region?
Please explain PAM vs. BLOSUM matrices in detail. Reading and lecture did not help clarify.
I would like to go through again how the scoring of each position in the matrix is done in dynamic programming (the matrix fill/scoring step).
1. How can one tell if it is convergent or homolog by sequence alignments?
2. Please explain the slide about global (Needleman-Wunch) vs local alignment (Smith waterman) again.
3. What is the name of the scoring system/ matrix? I couldn’t catch that in the video.
1. Is there a threshold for homology? At what point of similarity can you classify two proteins as homlogs?
2. I did not really understand the matrix scoring system for alignments either, could you please explain that further?